
Contact Information
Dept. of chemistry
and Biochemistry
University of
North Carolina
601 S. College Road
Wilmington, NC 28403
Office: DO 230B
Phone: 910-962-2439
e-mail: leehs[AT]uncw.edu

The Research Scope of Lee Group
Development and application of molecular dynamicsMolecular dynamics studies of lipid bilayers interacting with mutant peptides
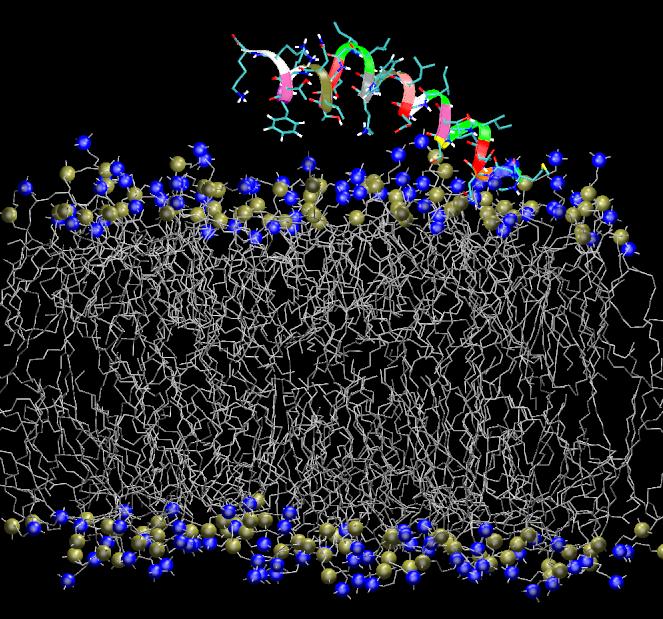
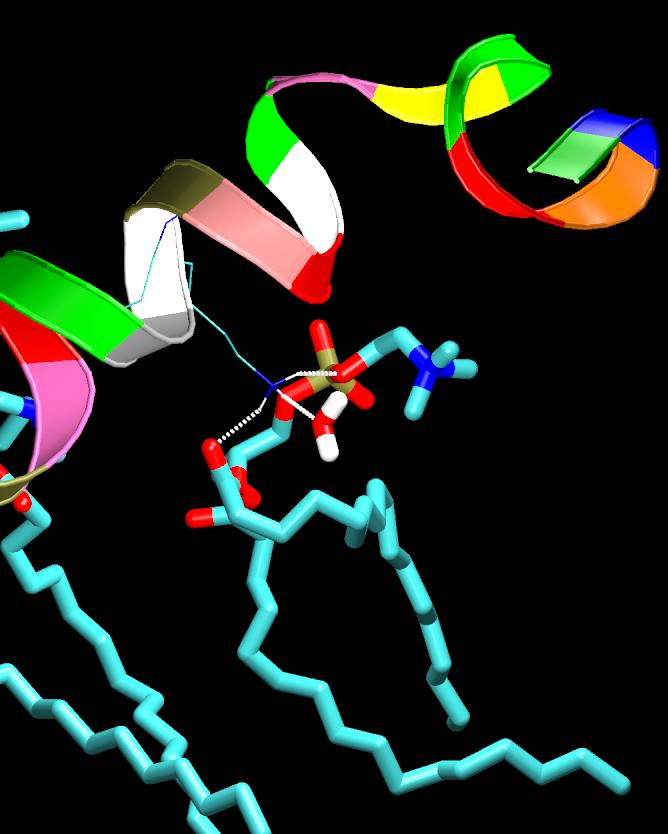
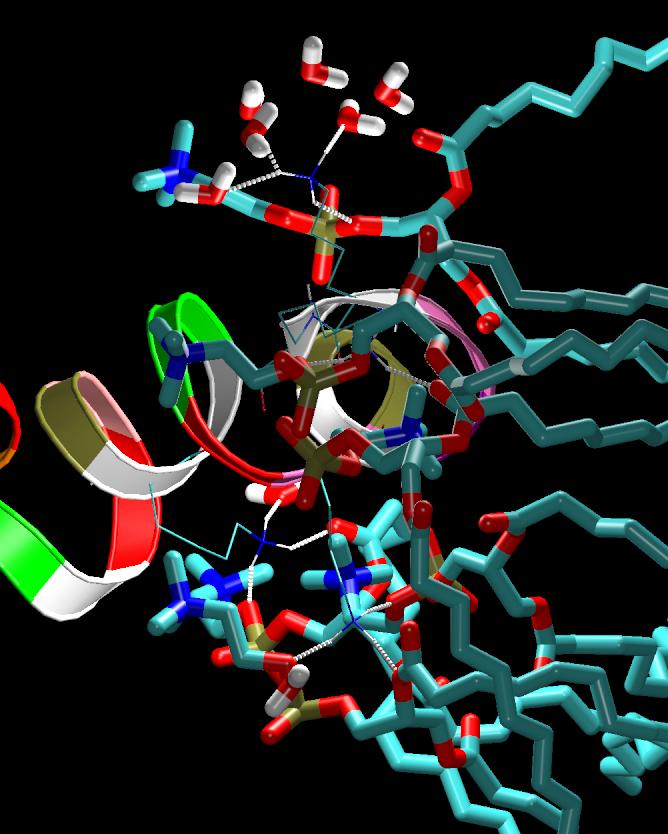
Antimicrobial, cytolytic peptides are amphipathic peptides that bind to cell membrane. They are known to be an important part of defense mechanism in eukaryotic organism. Although numerous mechanisms of membrane penetration by antimicrobial peptides have been proposed, no apparent peptide sequence has been found responsible for specific functions of peptides. One of the proposed membrane disruption mechanisms is "sinking raft" model, which suggests that cell-penetration mechanism and efficiency are directly related to the Gibbs free energy of insertion into the bilayer: If free energy change is smaller than ~20kcal/mol, peptides disrupt the bilayer by translocating accross the membranes. On the other hand, the peptides that require more than ~20kcal/mol of free energy change cannot translocate, but they will accumulate on the membrane surface until a pore forms. To investigate the validity of such hypothesis, molecular dynamics (MD) simulations are currently underway for the POPC lipid bilayers interacting with various peptides with designed sequences. These mutant peptides are designed to convert translocating peptides into non-translocating peptides visa versa. This project is in collaboration with Drs. Almeida at UNCW.
Catalytic effect of platinum doped carbon nanotube (Pt/CNT)
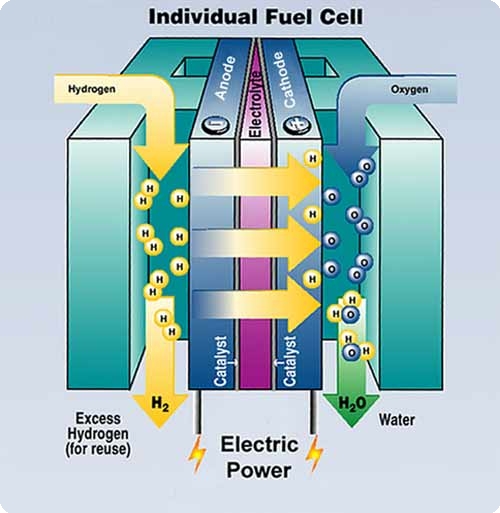
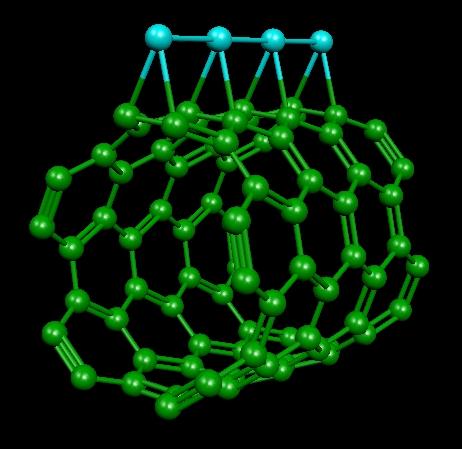
(picture from DOE website)
The proton exchange membrane fuel cell (PEMFC) is one of the most tantalizing future energy technology. However, wide-spread use of fuel cells has been hampered by the high cost of metal catalyst. To develop cost-effective PEMFC systems, it is highly desirable to maximize the efficiency of expensive Pt catalyst. Recently, a number of research groups have used Pt-doped carbon nanotube (CNT) as an alternative catalyzing/supporting material in PEMFC electrodes and reported superior performance of Pt/CNT electrodes over that of conventional carbon black based system. In this project, density functional theory (DFT) electronic structure calculations and ab initio molecular dynamics (AIMD) simulations will be used to charaterize the nature of Pt-CNT interaction and understand the catalytic role of Pt/CNT. In particular, we are investigating the oxidation/reduction mechanisms involving platinum clusters via nudeged elastic band (NEB) method.
Fluorescent PET (photoinduced electron transfer) sensors
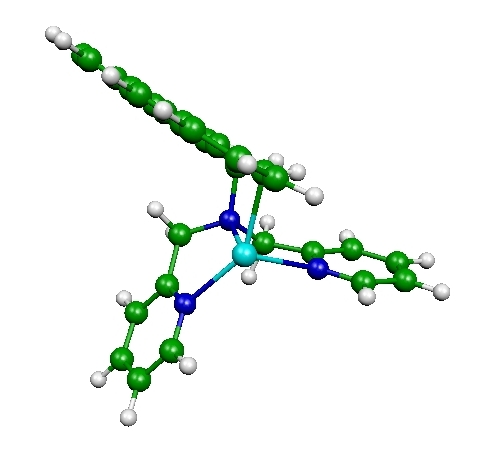
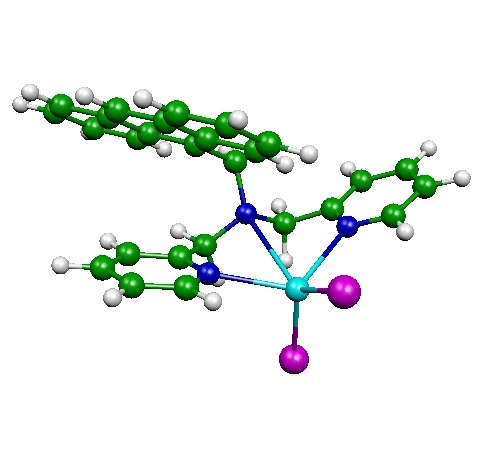
Ion sensors are of great interest to many researchers, including clinical biochemists and environmental scientists. In particular, fluorescent sensors based PET have been recognized as one of the most sensitive and practical ways to detect a wide variety of cations. Fluorescent sensors (or fluoroionophores) are made of two parts: fluorophore, which emits fluoresence light upon excitation, and a ligand or receptor, which binds to a cation. Without a cation, the fluorescence from the fluorophore is mostly quenched due to the electron transfer from the receptor to the fluorophore. When the receptor binds to a cation, such electron transfer is suppressed and fluoroscence signal increases drastically. Our interest is to develop similar type of fluorescent PET sensors for anions. In collaboration with Dr. Hancock, we investigate the structure and photochemical properties of fluoroionophores (an example shown above) with and without anions. This project involves time-dependent DFT (TDDFT) calculations for the fluoresence intensity as well as DFT calculations for the optimized structures of proposed sensors.
Ab initio molecula dynamics (AIMD) based on real-space basis
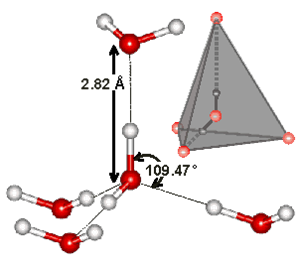
Currently, AIMD simulations are most commonly performed in plane-wave basis set. Indeed, the most popular AIMD code in the community, CPMD, is based on this technique. However, localized basis set techniques have been increasingly popular in recent years for improving the scaling with respect to the system size. Recently, a discrete variable representation (DVR) based approach was introduced as an alternative real-space method for the electronic structure calculation in AIMD simulations (see paper #20). DVR basis has excellent convergence properties, can be used in linear scaling approach and can be easily adapted for massively parallel applications. Car-Parrinello AIMD based on DVR basis was used in our recent study of neat water (see paper #21 and #22) and protonated water (see paper #27), which showed a significant improvement on structural and dynamical properties. This method was recently extended to non-periodic (cluster) system as well (see paper #24). We are currently extending the AIMD/DVR approach to path integral simulation for full nuclear quantum dynamics, as well as simulations with 1D (wire) and 2D (surface) periodic boundary conditiions.